文章摘要
【关 键 词】 蛋白质折叠、Transformer、生成模型、SimpleFold、结构预测
蛋白质的功能很大程度上由其精确的三维空间结构决定,而预测蛋白质如何从氨基酸链折叠成立体形态一直是生物学领域的核心难题。从1972年诺贝尔奖得主Christian Anfinsen的奠基性工作,到2020年AlphaFold2的革命性突破,这一领域经历了从实验方法到计算预测的漫长演进。AlphaFold2通过复杂特化的架构设计,如多序列比对和三角形更新模块,实现了与实验方法相媲美的预测精度,成为行业标杆。
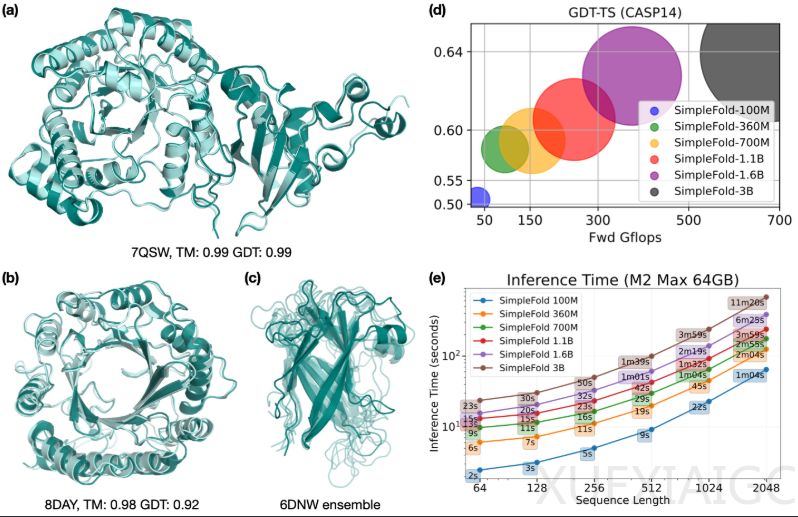
然而,苹果公司研究人员提出的SimpleFold模型挑战了这一范式。该研究证明,蛋白质折叠预测可能不需要复杂领域专用模块,仅需通用Transformer架构就能达到同等效果。SimpleFold将蛋白质折叠视为条件生成任务,采用”流匹配”技术,从高斯噪声出发逐步生成精确结构。其架构分为原子编码器、残基主干和原子解码器三部分,完全摒弃了AlphaFold2的特制组件,仅依赖基础Transformer模块和预训练蛋白质语言模型ESM2-3B的序列嵌入。
模型训练采用了前所未有的数据规模,混合PDB实验结构、AlphaFold预测结构和AFESM数据库近900万个代表性结构。在CAMEO22和CASP14基准测试中,SimpleFold-3B表现优异,达到AlphaFold2性能的95%以上,甚至在某些指标上超越同类模型。更重要的是,作为生成模型,SimpleFold能预测蛋白质的多种构象,在Apo/holo数据集上取得当前最佳性能,这对理解蛋白质动态功能和药物研发具有重要意义。
研究揭示了规模效应的强大作用:随着模型参数和数据量的增加,性能持续提升。这表明通用架构设计具有广阔发展前景,未来通过更大模型和更多数据,可能实现更优预测效果。这一发现为蛋白质结构预测领域提供了新思路,证明简洁通用的方法同样能解决复杂生物学问题,甚至可能超越特化设计的系统。该成果也体现了生成模型在科学计算领域的巨大潜力,为其他复杂系统的建模提供了有益借鉴。
原文和模型
【原文链接】 阅读原文 [ 2213字 | 9分钟 ]
【原文作者】 AIGC开放社区
【摘要模型】 deepseek/deepseek-v3-0324
【摘要评分】 ★★★★☆


相关文章


